O termo neurofibromatose se refere a duas desordens genéticas distintas, a neurofibromatose tipo 1 e a neurofibromatose tipo 2. A neurofibromatose tipo 1 é também conhecida como Neurofibromatose de Von Recklinghausen ou Neurofibromatose Periférica. A neurofibromatose tipo 2 é também conhecida como Neurofibromatose do Neurinoma do Acustico Bilateral ou Neurofibromatose Central. Ambas provocam a formação de tumores ao redor dos nervos.
Tem incidência de 1 em 3.500 nativivos e é uma doença autossômica dominante com penetrância completa. Além disso, apresenta expressividade variável, entre diferentes famílias, entre diferentes indivíduos de uma mesma família e entre diferentes segmentos de um mesmo indivíduo.
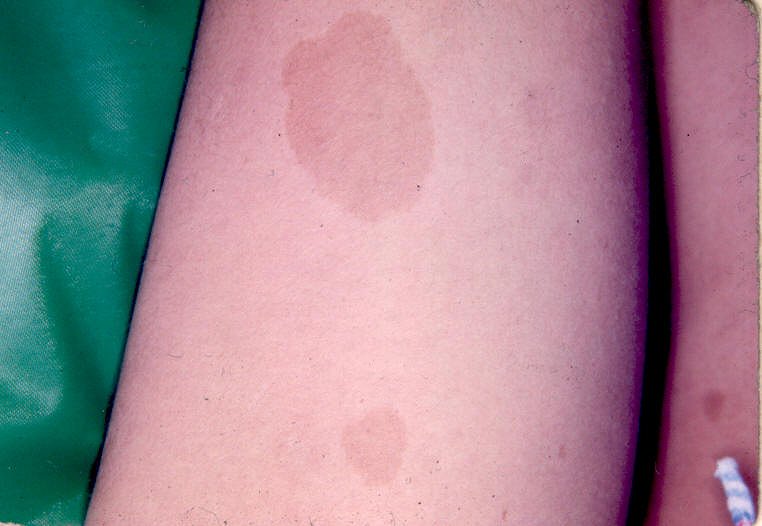
Entre as manifestações clínicas principais estão as manchas ovóides de cor uniforme café-com-leite e margens bem definidas; e as sardas, principalmente inguinais e axilares, que têm entre 1mm e 3mm de diâmetro e ocorrem agrupadas geralmente em regiões de superposição de pele. Os neurofibromas são tumores da bainha dos nervos periféricos e podem ser discretos ou plexiformes.
Os neurofibromas discretos são massas focais bem definidas que normalmente aparecem na puberdade e podem estar associados a prurido. Podem ser classificados como cutâneos ou subcutâneos. Os plexiformes envolvem múltiplos fascículos ou ramos de nervos e podem crescer exageradamente, apresentando degeneração maligna, tumores malignos da bainha dos nervos periféricos – TMBNP, que podem ser classificados como difusos ou nodulares.
A neurofibromatose afeta ambos os sexos e raças na mesma proporção. A neurofibromatose tipo 1 é a mais comum e afeta 1 a cada 4.000 pessoas, já a neurofibromatose tipo 2 é mais rara e afeta 1 a cada 40.000 pessoas.
Já estão disponíveis testes de DNA para portadores de neurofibromatose, porém o diagnóstico normalmente é feito através dos sinais clínicos. Os testes são geralmente usados para identificar se uma criança sem ou com pouco sinais é portadora de neurofibromatose, sendo importante na neurofibromatose tipo 2 visto que a desordem se manifesta tardiamente.
Recomenda-se aos afetados que se submetam periodicamente (uma vez por ano) aos seguintes exames: audiometria de ambos os ouvidos, exame oftalmológico (campos visuais), tomada de pressão arterial e exame da coluna vertebral. A maioria dos afetados leva uma vida praticamente normal e bem adaptada, uma vez que esses exames previnem complicações, que felizmente não são muito comuns.
Os exames devem sempre ser realizados por recomendação de um médico.
Entenda Melhor
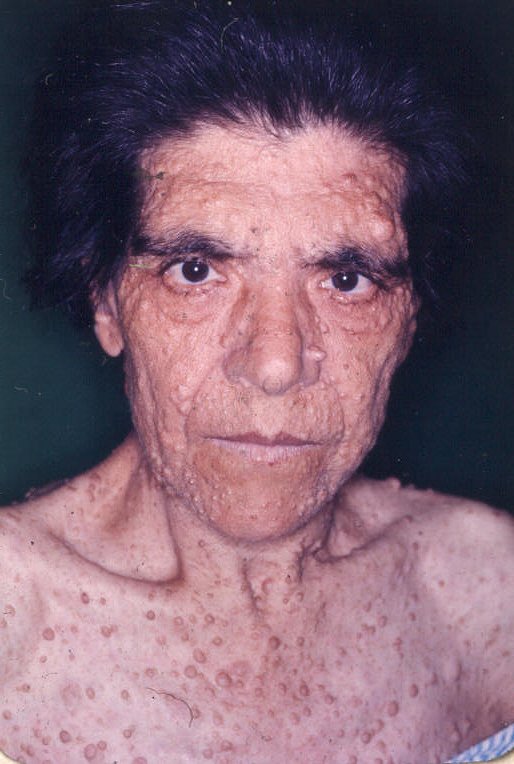
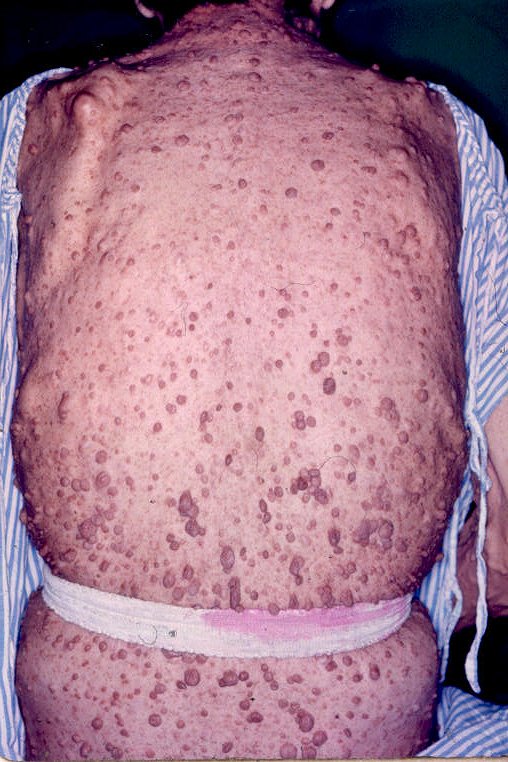
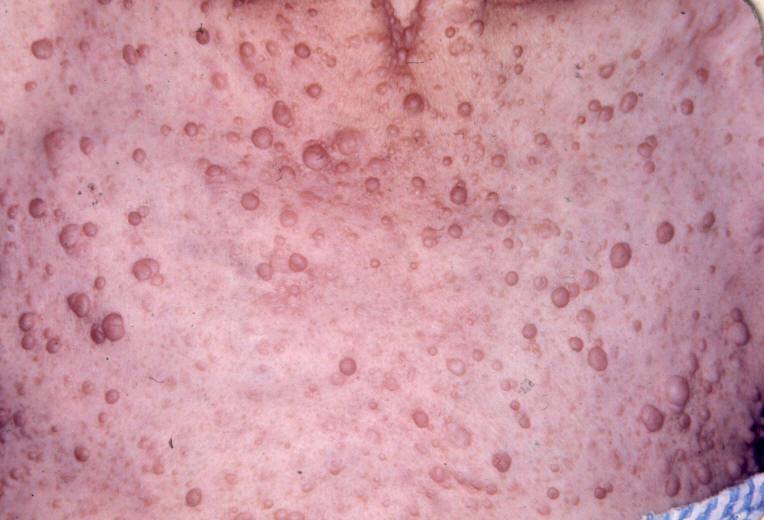

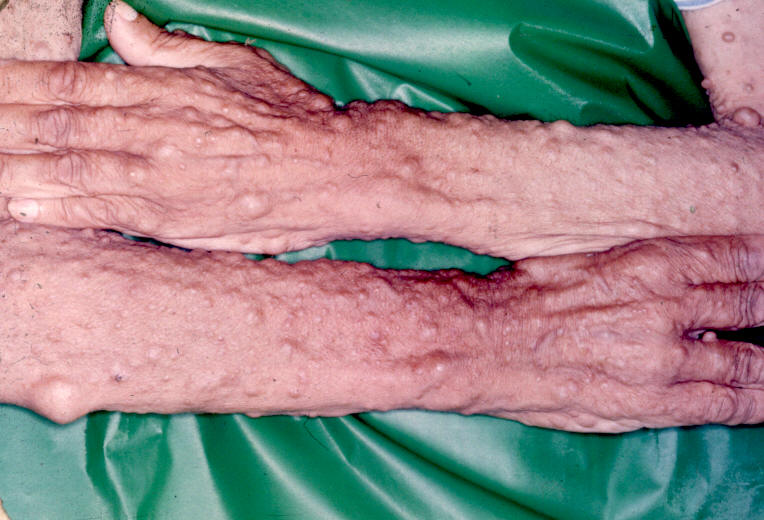
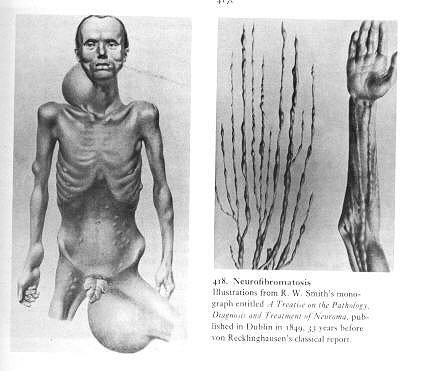
Ilustrações de uma monografia publicada em Dublin, por R. W. Smith em 1849, 33 anos antes do trabalho de von Recklinghausen.
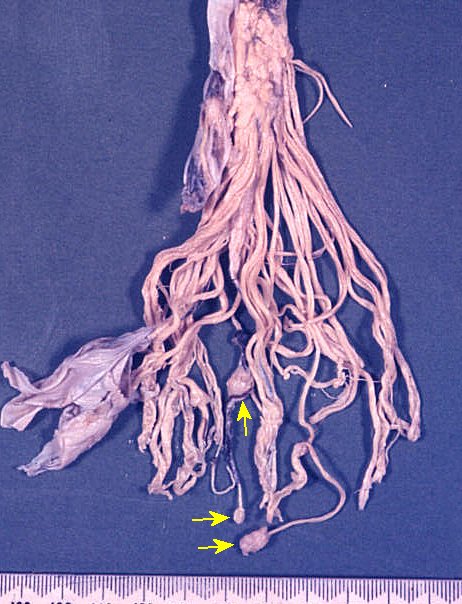
Cauda eqüina de paciente com neurofibromatose do tipo 2. Vários pequenos schwannomas aparecem como nódulos associados às raízes.
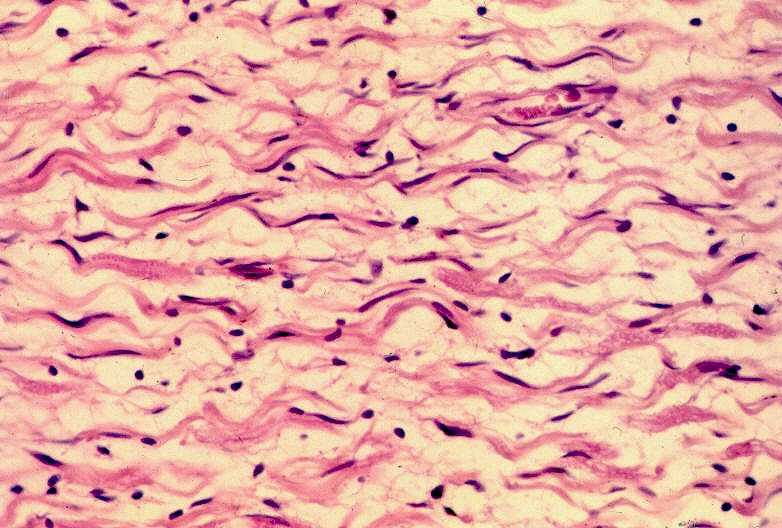
Detalhe de um neurofibroma corado por HE. As células são fusiformes e onduladas, em meio a matriz frouxa e pálida.
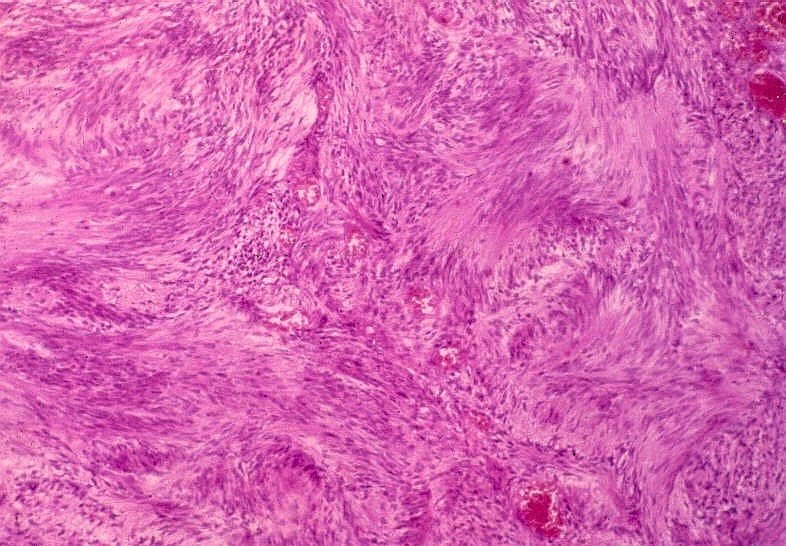
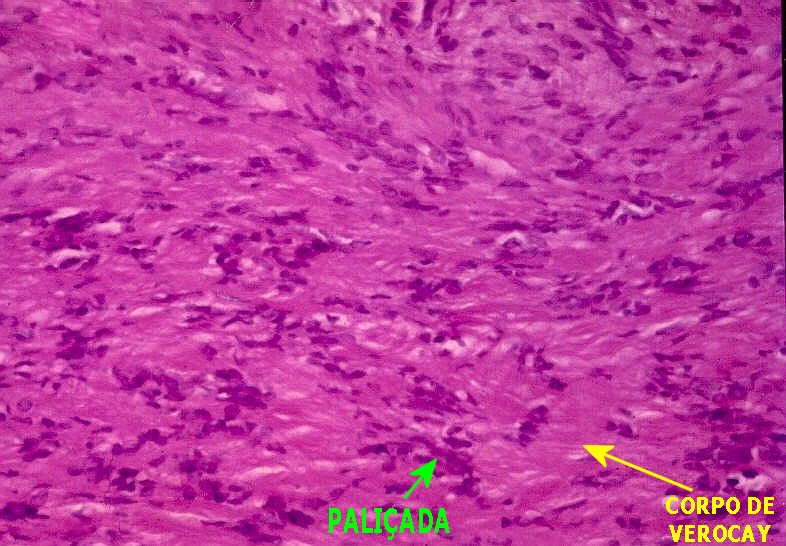
Os schwannomas são constituídos por células alongadas arranjadas em feixes compactos.
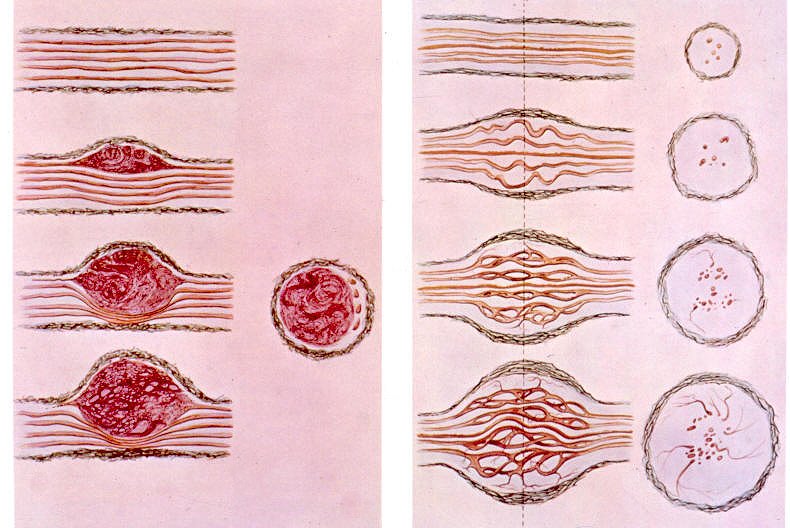
Comparação entre um schwannoma (fig à E) e um neurofibroma. O schwannoma cresce deslocando os axônios do nervo e no tumor propriamente dito não há axônios. Axônios podem ser observados na periferia do tumor ou na cápsula. Já no neurofibroma as células neoplásicas proliferam entre os axônios, divulsionando-os.
Outras alterações neurológicas incluem déficit cognitivo, alterações de coordenação motora, alterações de comportamento e/ou psiquiátricas, cefaléia, epilepsia, estenose de aqueduto, alterações cerebrovasculares e OBNIS – objetos brilhantes não identificados, que aparecem na ressonância magnética de encéfalo, sem apresentar efeito de massa ou realce após o contraste. Alterações esqueléticas podem incluir também: baixa estatura, macrocefalia, genu varum/valgum, deformidade esternal, displasias vertebrais, displasia da asa do esfenóide e displasias craniofaciais.
Entre as alterações cardiológicas estão hipertensão arterial sistêmica idiopática ou relacionada com feocromocitoma ou estenose da artéria renal por neurofibroma, cardiopatias congênitas e vasculopatias. As alterações endócrinas mais comuns são as puberais (puberdade precoce ou tardia) e a exacerbação da doença durante a gravidez.
A NF1 é uma síndrome de predisposição hereditária ao câncer e entre as neoplasias mais comuns estão os tumores malignos da bainha dos nervos periféricos – TMBNP, que devem ser suspeitados quando há dor ou alteração de tamanho ou consistência de um neurofibroma plexiforme. O risco cumulativo para um portador, durante sua vida, de desenvolver TMBNP é de 8% a 13%. O tratamento é cirúrgico.
Os gliomas não-ópticos, quando ocorrem, têm risco de malignização maior do que o da população em geral. Também é comum a ocorrência de neoplasias hematológicas, principalmente leucemias e linfomas não-Hodgkin. Os feocromocitomas têm forte associação com NF1, geralmente acometem as adrenais de indivíduos adultos e são bilaterais em 10% dos casos e malignos em 10%. Podem causar HAS, sudorese, taquicardia, vertigens. O aumento da epinefrina está associado à HAS intermitente e ao aumento de norepinefrina. Outros tumores que ocorrem associados à NF1 são carcinóides, rabdomiossarcomas, tumores da crista neural e tumor de Wilms.
O gene da neurofibromatose está no cromossomo 17q11.2, tem 60 Exons numerados de 1 a 49, sendo que 3 deles - 9, 23a e 48a - apresentam splicing alternativo. No Intron 27b, encontram-se 3 genes transcritos na direção oposta do gene NF1, EV12A, EV12B, OMGP. O NF1 tem alta taxa de mutações novas e existem vários pseudogenes, em outros cromossomos.
O mRNA é expresso em todos os tecidos com três isoformas: a primeira com o Exon 23a incluso, associada à diminuição da atividade catalítica enzimática e não encontrada nos feocromocitomas; a segunda com o Exon 48a, encontrada nos tecidos musculares e a terceira com o Exon 9a incluso, encontrada no cérebro durante a embriogênese.
A proteína neurofibromina tem 2.818 aminoácidos, 327 kDa e atua em vias de proliferação e sinalização celular. Apresenta uma região de homologia com a família de proteínas ativadoras de GTPase (GAP), com o domínio relacionado a GAPs (GRD) e atua na regulação de ras, proto-oncogene que, quando ativo, atua no crescimento celular e controle da apoptose. Com a neurofibromina truncada, ras não é inativado e há desregulação no crescimento celular.
Mutações foram encontradas no gene NF1 em pacientes com neurofibromatose por meio de várias metodologias. A análise do DNA, domínio GRD, permite a detecção de 20% de mutações e a análise da proteína por PTT – teste da proteína truncada – detecta 70% das alterações, confirmadas posteriormente por seqüenciamento direto. Já foram descritos vários tipos de alterações: alterações cromossômicas, deleções de todo o gene, vários Exons ou microdeleções, inserções e mutações de ponto, geralmente restritas à família. A mais recorrente delas é a C5839T no Exon 31. A maior parte das deleções intragênicas tem origem materna e as mutações de ponto, origem paterna. Poucas correlações entre o genótipo e o fenótipo existem: as microdeleções parecem aumentar o risco para TMBNPs e grandes deleções estão associadas a um fenótipo mais grave, talvez por envolver outros genes. Nos tumores, foi verificada a inativação do alelo normal, por perda de heterozigosidade, o que levou à classificação do gene NF1 como um gene supressor de tumor.
O acompanhamento de um paciente com NF1 deve incluir história familiar, avaliação oftalmológica, radiografia de esqueleto, ultrassonografia de abdome e, em casos especiais, tomografia computadorizada de crânio e triagem para feocromocitoma. Deve ser anual para adultos e semestral para crianças, realizado por uma equipe multidisciplinar familiarizada com a história natural da doença.
Existem poucos estudos clínicos para tratamento. Estudos em fase I de lesões sintomáticas foram feitos com talidomida, com redução do tamanho dos neurofibromas em 1/3 dos casos e melhora dos sintomas em 1/3; inibidores orais da farnesil transferase, sem redução do tamanho dos neurofibromas; e estudos clínicos fase II foram realizados com fumarato de cetotifeno, sem redução do tamanho dos neurofibromas e com melhora dos sintomas em alguns pacientes; e ácido retinóico ou interferon alfa, com melhora sintomática em 14% dos casos e redução em 8% dos casos.
Trechos Biográficos de:
Carey JC, Viskochil DH. Neurofibromatosis type 1: a model condition for the study of the molecular basis of variable expressivity in human disorders. Am J Med Genet 1999;89:7-13.
Korf BR Malignancy in neurofibromatosis type 1. The oncologist 2000;5:477-85.
Dasgupta B, Gutmann DH Neurofibromatosis 1: closing the GAP between mice and men. Curr Op Gen Dev 2003;13:20-7.
Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51-7.
Nenhum comentário:
Postar um comentário